
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Detecting Alternate Translation Initiation Events (2020/11/27)
This week on Twitter I made a few suggestions about things to keep in mind when trying to modify a search engine so that it could identify peptides generated by alternative translation initiation events. This type of initiation is fairly
common in mammals: it is the result of "leaky-scanning" of mRNA by the 40S ribosomal subunit prior to translation. It is a well-understood mechanism
, but it has not been generally applied in proteomics data analysis. For details about the mRNA mechanism, see Alan G. Hinnebusch, 2011 and Marilyn Kozak, 2002.

If you are interested in adding an algorithm to an existing search engine to find alternate translation initiation sites, you should remember the following tips: 1/9
Almost all alternate initiation sites are the 2nd M in a protein sequence; 2/9
Alternate initiation M's are subject to the same co-translational modifications as M1 initiation: acetylation and/or removal of the M and acetylation of the following residue; and 3/9
Peptides beginning at the alternate M are tryptic peptides like any other. 4/9
If you want some data to test your new algorithm (or maybe an open search will find them?), try the "Keratinocyte" data from PXD019909. It has multiple observations of peptides generated by this mechanism, e.g.: 5/9
s 55 MDGAGAEEVLAPLR 68 l from GARS1:p, with M55+acetyl and M55+oxidation; 6/9
m 9 ADKMDMSLDDIIK 21 l from ALYREF:p, with a 1 missed tryptic cleavage (K10) and several potential methionine oxidations; 7/9
m 33 STASVEIDDALYSR 46 q from UBA6:p, with S33+acetyl. 8/9
About 5% of peptides observed from translation initiation events should correspond to these "leaky-scanning" peptides. /fin
Added note: 5% only holds for cellular proteins. Most extracellular proteins do not have peptides from translation initiation events, at either M1 or alternate sites.
Adversarial Analysis (2020/09/30)
This week on Twitter I mentioned the idea of applying an approach I refer to as "adversarial analysis" to data. This concept is a way of seeing experimental data in an atypical manner; treating the data as an instrument for proving that the proposed experiment was really performed as designed. It can be quite challenging, but once you get the idea it becomes difficult to stop applying it to every new data set you run across.
What I call "adversarial analysis" is an attempt to deal with the fact that what actually happens in a lab is imperfectly recorded in written experimental methods (sometimes very imperfectly). /1
Rather than accepting any part of a method (reagents, chromatography, MS/MS conditions, derivatizations, etc) as being true, I try to come up with the simplest way to test the data that will show the extent to which it was false. /2
These "best ways" tend to be indirect: using some property that can be derived from the results that was not directly measured or controlled for during the experiment. /3
Many people do this sort of thing, but because I only deal with other people's data, I need to do it more comprehensively than most. It is more of a mind set than a routine set of steps. /…
For example, if the method for a data set states that the sulfhydryl groups of cysteine residues have been blocked using iodoacetamide, it would seem natural to assume that all of the cysteines now have an S-H3C2NO rather than the original S-H. Applying adversarial analysis to this assertion requires the data to be tested assuming that the derivatization occurred correctly, but also to check for other common derivatizations (acrylamide, β-mercaptoethanol, cystine, glutathione, trioxidation) as well as unmodified cysteine. This check need not be run on every LC/MS/MS data file in a large study: simply test a few selected in a block-random manner appropriate for the overall experimental design.
Field's Law (2020/09/15)
 This week on Twitter I mentioned a simple principle that has come in handy time-and-time again. When I worked at Rockefeller University in the late '80's, I joined Dr. Frank Field's lab—the inventor of chemical ionization—under Brian Chait (who took over the lab when Dr. Field retired). I wanted to select a good protein to use as an exemplar test material for the MALDI source I was building, so I asked Dr. Field's advice about it. During the discussion, he formulated what I now call Field's Law.
This week on Twitter I mentioned a simple principle that has come in handy time-and-time again. When I worked at Rockefeller University in the late '80's, I joined Dr. Frank Field's lab—the inventor of chemical ionization—under Brian Chait (who took over the lab when Dr. Field retired). I wanted to select a good protein to use as an exemplar test material for the MALDI source I was building, so I asked Dr. Field's advice about it. During the discussion, he formulated what I now call Field's Law.
While only known to people who had the good fortune to work in Frank Field's lab, Field's Law states: There is no such thing as an average protein.
At the time it seemed to be an odd thing to say, but it was true and has broad, general implications for how one should think about proteomics, particularly for researchers that come from a physical science background. In most physical sciences, one often tries to estimate the outcome of a proposed experiment by considering the behavior of the system "on average". This phrase implies that while there will be some variablity in the system, assuming that the variation is normally distributed will allow you to make a reasonable estimate: at worst, to within an order of magnitude. When dealing with the properties of a collection of proteins isolated from an organism, this assumption of "average" behavior is never true and always leads to expectations that differ from observations.
One of the phenomena that contributes to this property of proteins is that in many dimensions proteins cannot be modeled with a Gaussian distribution. Instead, they frequently require a heavy-tailed distribution for any type of realistic modeling. This type of distribution falls off slower than exponentially, which has the effect of rendering "mean" values much less useful as approximations.
Another reason is that very little about proteins is stochastic, from their concentrations to their molecular compositions. They are purpose-built devices that are under strict quality control and if anything about them deviates from their original design, they are quickly discarded.
The effects of this non-normal behavior can be seen in many proteome-wide statistics. For example, if one draws a histogram of the number of proteins vs the number of residues in a protein for a given proteome, it results in the following curve (in this case for R. norvegicus):
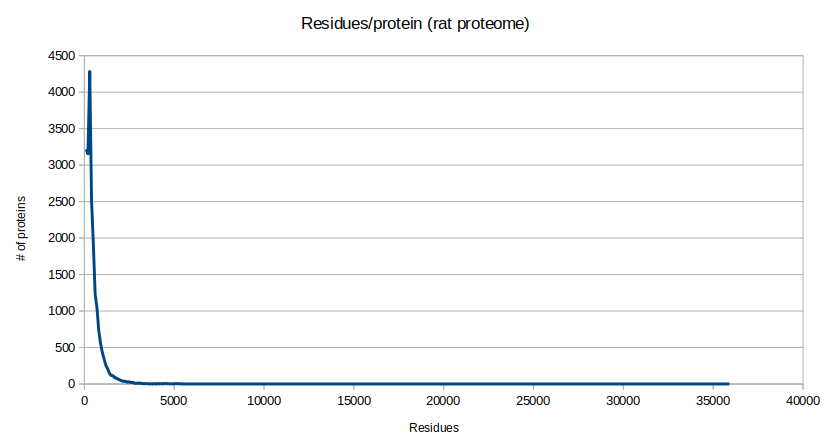
The usual statistical parameters that are valuable for describing a Gaussian curve (mean, mode, median, standard deviation) are not useful parameters for describing this type of long-tailed distribution. Therefore, any discussion using a statement such as:
So if you are ever discussing proteins and any word that means "average" comes to mind, think of the stern looking guy in the bow tie saying "no".
Drawing an annotated spectrum from a single scan (2020/09/09)
I asked a question on Twitter about how to generate a graph of a single scan extracted from a Thermo
.raw file and annotate the graph with a known peptide sequence assignment using stand-alone open source software. There were quite a few responses, but there did not seem to be anything available to perform this specific task.
Is there any stand-alone open source software that allows you to take:1. a .raw file; 2. an MS/MS scan number; & 3. a peptide sequence + mods and from that generate a nice looking spectrum with matched ions marked up.
Once it became clear there wasn't really anything available, writing something to do this became the most useful option. Python 3 was chosen as the programming language and a set of methods + some demo software was constructed and made available on GitHub, under the name scan_graph. The project uses pymsfilereader to access the Thermo .raw file. The demo will generate a PNG showing an MS/MS spectrum with the assigned peaks marked with colors, similar to the example below.
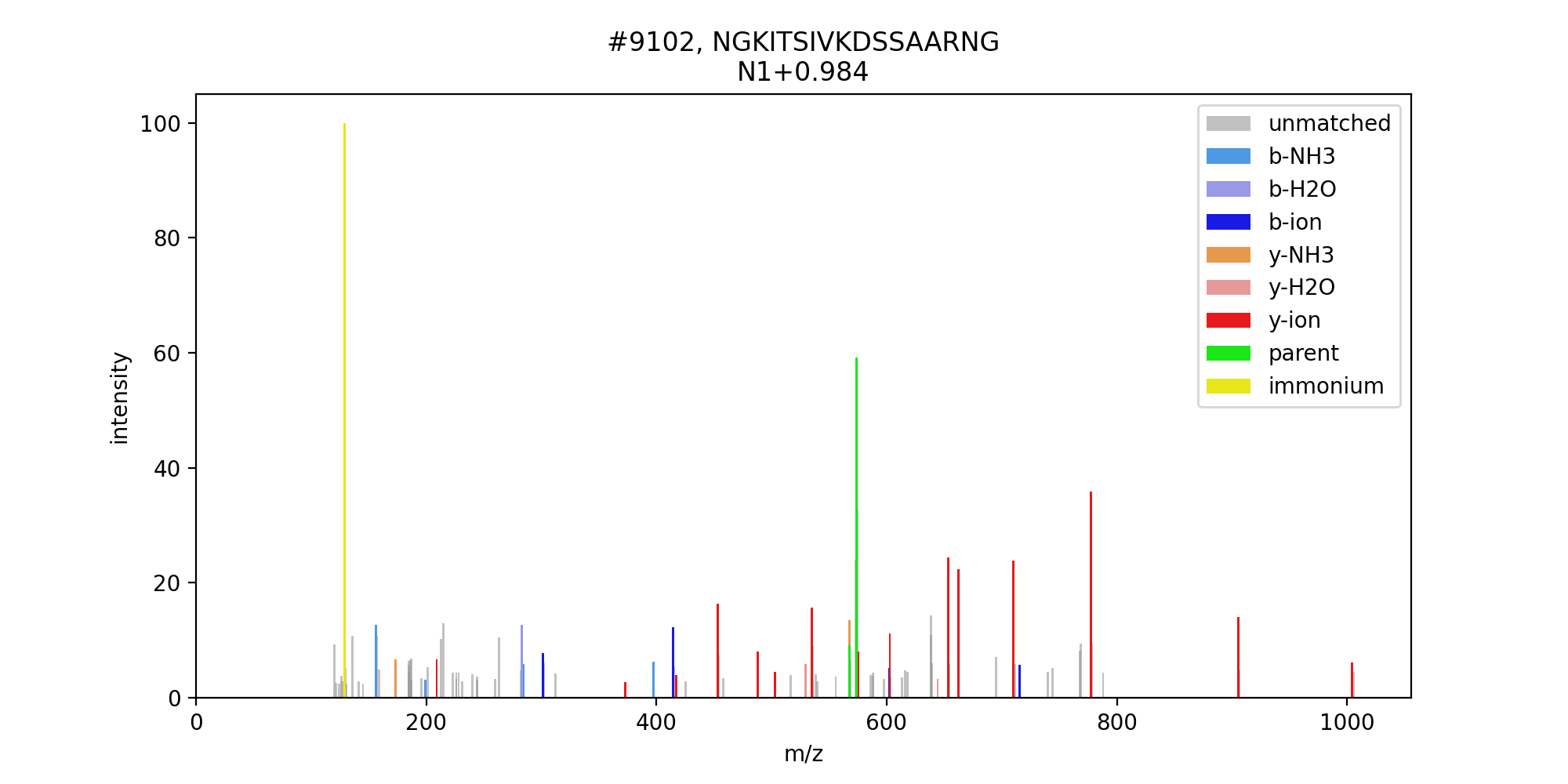
Please note that this software does not generate an intermediate text file (e.g., mzML or MGF format): it extracts the MS/MS spectrum directly from the .raw file based on the scan number.
Terminology for multiple PTMs on the same acceptor site (2020/09/01)
Proteomics has a significant problem with terminology. Even though it has been around as a field since
the mid-1990s, it has not developed simple systematic terms for referring to protein phenomena that proteomics has
discovered. Instead, terms for similar phenomena have been introduced in papers that are adequate for the
purposes of a particular manuscript, but which have failed to be adopted by the community at large. Even well-read,
experienced researchers can struggle to apply these orphaned terms to their own research results or in theoretical discussions.
Yesterday a Twitter thread
was started for community input that resulted in some interesting discussion. The thread started with the following question regarding post-translational modifications (PTMs):
Is there a term (or terms) for the phenomenon of multiple PTMs possibly occupying the same protein residue? For example, a lysine that may be either acetylated, SUMOylated or ubiquitinylated.
This particular issue is not a new one: the first discussion of the observation was probably associated with eukaryote histone PTMs, where it was known that changes in the PTMs occupying particular acceptor sites was associated with changes in chromatin structure.
A group of interested scientists responded with useful suggestions. The responses clearly demonstrated that there is no
established terminology for this commonly observed phenomenon, although many papers have made reference to it. The suggested terms fell into two general groups: those that implied a mechanism or function and those that were neutral regarding the phenomenon.
The mechanistic/functional suggestions for this type of PTM were as follows:
As a starting point for further discussion, I suggest adopting the neutral terms alternative PTM (abbreviation: aPTM, plural aPTMs) for each PTM associated
with a particular site and SAM for the acceptor itself. For example: "acetylation and ubiqutinylation are aPTMs at 17 SAMs in human ribophorin 1".
Microheterogeneity has such a well-established meaning
in the protein glycosylation community that using it for acetylations or methylations may be confusing for many readers. Instead, I would suggest describing carbohydrate microheterogeneity at a SAM as a type of aPTM.
Each of the mechanistic/functional terms can be accommodated as special cases of aPTMs for any specific SAM, as warranted by the evidence.
If you wish to participate in the ongoing Twitter discussion of this topic, please add the hash tag "#altptm" to your tweets.
Beta release of the PSM assignment app idX v.2 (2019/11/26)
 Over the past few months, there has been an effort made to develop simplified, open-source PSM assignment
software that makes use of the built-in, rapid associative array mechanisms that are now common-place in
many computer languages, e.g., "dict" and "set" in Python, "hash"
and "set" in Perl, or "map" and "set" in C++.
Over the past few months, there has been an effort made to develop simplified, open-source PSM assignment
software that makes use of the built-in, rapid associative array mechanisms that are now common-place in
many computer languages, e.g., "dict" and "set" in Python, "hash"
and "set" in Perl, or "map" and "set" in C++.
The first attempt
at this was SE, mentioned below. This approach had several problems, most
notably a slow down due to excess memory usage.
A re-think of the problem lead to another implemention of the idea using more memory efficent object structures to store
and retrieve spectrum and peptide kernel information. This new implementation, idX, performed much better
than SE in terms of memory usage and PSM assignment speed. That being said, it still used enough memory that it would
be difficult to run multiple PSM assign jobs on the same device unless it had more memory than is commonly installed
on commodity computers.
While experimenting with a C++14 port of the idX project (originally written in Python3), it became clear that
there was a much more memory efficient set of objects for storing both the spectrum and kernel information for the
PSM assignment process, based on simple pairs of mass and intensity:
in STL C++, pair<int64_t,int64_t>. Using this approach greatly simplified the code and reduced the PSM assignment problem to a matter of looking up the pairs associated with a particular peptide in a map of pairs associated with a spectrum. It also reduced the amount of memory used to an amount that would not require special hardware for execution.
The code for this new approach (idX v.2) is publicly available.
A simplified web interface that can be used to test the approach is also available online.
If you would like to convert a Thermo RAW file to an MGF using Proteome Wizard's msconvert utility, the suggested
settings are:
>msconvert FILE.raw --mgf --filter "peakPicking true 2" --filter "msLevel 2"
Succinylation artefact & NHS-ester quantitation reagents (2019/6/22)
 Many of the reagents commonly used for MS-based proteomics quantitation are based on N-hydroxysuccinimide esters
(illustrated here).
These reagents can be used to derivatize free amines (peptide N-terminii and lysine ε-amino groups) with a wide range
of moieties. Recently, it has been observed that a side reaction of this process — the formation of succinylated amines in
place of the desired derivative — can result in observable amounts of peptides in which the desired moeity has been replaced by succinylation (+100.016 Da).
This side reaction results in somewhere between 0–25% of identifiable PSMs. The number of succinylated PSMs is unevenly distributed across fractions of multidimensional
chromatography separations. Experimental groups are encouraged to try to minimize this reaction and any analysis of
the data resulting from this type of derivatization should try to estimate of the extent of this reaction.
Many of the reagents commonly used for MS-based proteomics quantitation are based on N-hydroxysuccinimide esters
(illustrated here).
These reagents can be used to derivatize free amines (peptide N-terminii and lysine ε-amino groups) with a wide range
of moieties. Recently, it has been observed that a side reaction of this process — the formation of succinylated amines in
place of the desired derivative — can result in observable amounts of peptides in which the desired moeity has been replaced by succinylation (+100.016 Da).
This side reaction results in somewhere between 0–25% of identifiable PSMs. The number of succinylated PSMs is unevenly distributed across fractions of multidimensional
chromatography separations. Experimental groups are encouraged to try to minimize this reaction and any analysis of
the data resulting from this type of derivatization should try to estimate of the extent of this reaction.
Notes:
Alpha release of the SE PSM assignment project (2019/5/12)
The SE PSM assignment software project is an experimental project to create a simple, but full-featured
proteomics MS/MS search engine written in Python 3. Information about the project and the code
is available at https://github.com/RonBeavis/SE/wiki.
Potential service disruptions (2019/3/4)
The GPMDB web servers are being moved to a new location this week. There will be some periods during which
various service may be unavailable while the servers are being physically moved and while the global
DNS system catches up with the new IP addresses associated with the corresponding URLs.
Added 2019/3/13: the server move and network reconfigurations are complete and the system
seems to be working properly. There were a few problems with the network proxy configuration
from Sunday until yesterday, but a new set of parameters seem to have cleared up those issues.
Does proteomics need another data file format (JSMS)? (2019/2/26)
Proteomics MS/MS data can be archived, transmitted and archived using many different non-proprietary file formats
(mzML, mzData, mzXML, MGF, DTA, etc.). They all have their strengths and weaknesses (as well as proponents and opponents).
The XML-based formats have become standards for the archiving of large data sets in a format that does not require
any special vendor-specific software to access.
These XML formats are very good for the reliable storage of data sets, but they tend to be quite large and parsing
out specific features from these files requires specific knowledge of the XML structure. Because of their size, transmitting
these files over a network can be quite time consuming.
We are in the process of developing a new file format based on the JSON Lines style:
a more recent development in text-based file formats. The goal is to provide a useful replacement for the MGF format, which is
commonly used to communicate MS/MS data between software APIs. The new format, tentatively referred to as JSMS, has a
project page and a GitHub repository.
If you are interested in working on this project, just let us know at the email address on the project page.
How to use a RESTful API so as not to emulate a DOS attack (2019/2/9)
 Many biomedical databases (including our own GPMDB and g2pDB) make available APIs that use a representational state transfer (REST) structure to
easily retrieve interesting slices of information using simple Web requests to generate results in a simple to parse text format, e.g., JSON.
These interfaces are meant to give application developers access to this information in a straightforward way that does not
require them to install anything: they can just make the requests using the HTTP libraries that are available in most current
programming languages.
Many biomedical databases (including our own GPMDB and g2pDB) make available APIs that use a representational state transfer (REST) structure to
easily retrieve interesting slices of information using simple Web requests to generate results in a simple to parse text format, e.g., JSON.
These interfaces are meant to give application developers access to this information in a straightforward way that does not
require them to install anything: they can just make the requests using the HTTP libraries that are available in most current
programming languages.
The simplicity of using these interfaces can lead to problems for the REST API provider for several reasons. These APIs may
be frequently used, but it is almost impossible to obtain on-going funding to operate or maintain them. Therefore they tend
to be operated on a shoestring, with very modest hardware resources being used to service the requests. Even very large
bioinformatics platforms are hesitant to devote significant computational resources to these APIs, preferring to use
their resources for new projects or higher profile aspects of ongoing projects.
If you wish to make responsible use these resources (and avoid getting blocked), you should keep in mind the fact that the
resource probably is less capable than you may think. I would make the following suggestions for how to access these APIs:
www.thegpm.org moved to a new server (2018/7/24)
The domain names thegpm.org and www.thegpm.org has been assigned to a new
server. The old server (which had hosted the sites for 7 years) is being shifted
to a new role.
Termination of publicly available searches (2018/6/16)
Starting last Saturday (June 9, 2018), GPM will not longer be making its proteomics
data analysis servers available to the public. This service had been made available
for the last 15 years, but the aggressive misuse of the site by some set of individuals
has made it impractical to continue operations. GPMDB will continue to be available.
Addition of HTTPS service for GPMDB (2018/4/19)
For the anyone interested in enhanced privacy in browsing, GPMDB is now available via encrypted HTTPS
in addition to the normal HTTP protocol. The two protocols should result in the same output, but
will help if you are concerned about people snooping into your data browsing habits. To access GPMDB via
HTTPS, just go to https://gpmdb.thegpm.org. You should
see a lock symbol of some type (similar to 🔒) on the left side of your browser's URL line. HTTP access
will continue to be allowed, but it will be discontinued June 1, 2017.
Note: as of April 22, this site (thegpm.org & www.thegpm.org) will be redirected to HTTPS by default. HTTP access will no longer be permitted for [www.]thegpm.org, effective immediately.
Interesting data: PXD006367 (2017/5/23)
 The raw data set was made available through the ProteomeXchange site,
PXD006367.
It was published as part of the study entitled
"Cross-Sectional Association of Salivary Proteins with Age, Sex, Body Mass Index, Smoking, and Education"
by
Murr A, Pink C, Hammer E, Michalik S, Dhople VM, Holtfreter B, Völker U, Kocher T and Gesell Salazar M,
published in
J Proteome Res. 2017 May 18
(PubMed).
These experiments were represented in GPMDB by
209 files, each an individual LC/MS/MS run.
The raw data set was made available through the ProteomeXchange site,
PXD006367.
It was published as part of the study entitled
"Cross-Sectional Association of Salivary Proteins with Age, Sex, Body Mass Index, Smoking, and Education"
by
Murr A, Pink C, Hammer E, Michalik S, Dhople VM, Holtfreter B, Völker U, Kocher T and Gesell Salazar M,
published in
J Proteome Res. 2017 May 18
(PubMed).
These experiments were represented in GPMDB by
209 files, each an individual LC/MS/MS run.
This study was made to identify the most abundant human proteins in saliva obtained from a large number of individuals with some
clinical classifications made of the people involved. Saliva is a rather challenging body fluid for reproducible proteomics
and the sample preparation method used here was designed to try to minimize the amount of variability associated with complex
sample preparation schemes. The results were all single HPLC-MS/MS analyses of saliva obtained from individuals. The manuscript discusses
the human proteins found in the saliva, but for some reason it is silent on the microbiome protein and species variability
found in the data (e.g., GPM06610007910 vs
GPM06610007907, filtered for microbiome proteins only).
All of the usual bacterial suspects are present: Neiseria, Streptococcus, Haemophilus, Prevotella, Aggregatibacter, Rothia, et cetera.
Anyone interested in the application of proteomics methods to microbiome characterization should take a close look at this data to
see which proteins are easily detectable and the degree of microbiome variability found in these sub-populations.
Interesting data: PXD001197 (2017/3/5)
 The raw data set was made available through the ProteomeXchange site,
PXD001197.
It was published as part of the study entitled
"Cellular Signature of SIL1 Depletion: Disease Pathogenesis due to Alterations in Protein Composition Beyond the ER Machinery"
by
Roos A, Kollipara L, Buchkremer S, Labisch T, Brauers E, Gatz C, Lentz C, Gerardo-Nava J, Weis J and Zahedi RP,
published in
Mol Neurobiol. 2016 Oct;53(8):5527-41
(PubMed).
These experiments were represented in GPMDB by
18 files, each an individual LC/MS/MS run.
The raw data set was made available through the ProteomeXchange site,
PXD001197.
It was published as part of the study entitled
"Cellular Signature of SIL1 Depletion: Disease Pathogenesis due to Alterations in Protein Composition Beyond the ER Machinery"
by
Roos A, Kollipara L, Buchkremer S, Labisch T, Brauers E, Gatz C, Lentz C, Gerardo-Nava J, Weis J and Zahedi RP,
published in
Mol Neurobiol. 2016 Oct;53(8):5527-41
(PubMed).
These experiments were represented in GPMDB by
18 files, each an individual LC/MS/MS run.
This data set is a good demonstration of what can be obtained by using label-free 1D HPLC/MS/MS to profile differences
induced in the common cell line HEK-293.
The study reliably identifies about 3,000 distinct protein groups per LC/MS/MS experiment from about 30,000 high quality
peptide-to-sequence matches (PSMs). The PSMs are remarkable in that there were very few experimental artifacts, allowing the
reliable detection of phosphorylations, acetylations and dimethyl-arginines, as well as a good distribution of the
SAVs commonly observed in HEK-293 cells. As is normal in HEK-293 cells, the E1B 55K and E1B 19K proteins
from Human mastadenovirus C are both prominently observed, e.g., 50 PSMs associated with these two protein
GPM64230001481.
Anyone interested in pushing the confidence limits in protein detection should consider using this data
set as an example of unusually good quality data from an hybrid linear quadrupole ion trap/orbitrap instrument.
Interesting data: MSV000079017 (2017/1/29)
 The raw data set was made available through the Massive ProteomeXchange site,
MSV000079017.
It was published as part of the study entitled
"Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells"
by
Krönke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, Svinkina T, Heckl D, Comer E, Li X, Ciarlo C, Hartman E, Munshi N, Schenone M, Schreiber SL, Carr SA, and Ebert BL,
published in
Science. 2014 Jan 17;343(6168):301-5
(PubMed).
These experiments were represented in GPMDB by
90 files, each an individual LC/MS/MS run.
The raw data set was made available through the Massive ProteomeXchange site,
MSV000079017.
It was published as part of the study entitled
"Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells"
by
Krönke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, Svinkina T, Heckl D, Comer E, Li X, Ciarlo C, Hartman E, Munshi N, Schenone M, Schreiber SL, Carr SA, and Ebert BL,
published in
Science. 2014 Jan 17;343(6168):301-5
(PubMed).
These experiments were represented in GPMDB by
90 files, each an individual LC/MS/MS run.
This data set is a great example of how well current methods work for isolating ubiquitinylated peptides. Many of
the analyses that target the lysine epsilon-amino-KK remnant result in more than 70% of the identified peptides corresponding
to this modification. The results clearly show the folly of the often-quoted canard about the incompatibility
of iodoacetimide cysteine blocking and ubiquitination detection: the problem only arises if the cysteine-blocking
reaction is done very poorly. Attempts to replace iodoacetamide with the less reactive chloroacetamide usually
result in an unacceptible loss of cysteine-containing peptides, which constitute 20% of observable tryptic peptides.
Interesting data: MSV000080368 (2017/1/14)
 The raw data set was made available through the Massive ProteomeXchange site,
MSV000080368.
It was published as part of the study entitled
"Proteomic Analysis of Pemphigus Autoantibodies Indicates a Larger, More Diverse, and More Dynamic Repertoire than Determined by B Cell Genetics"
by
Chen J, Zheng Q, Hammers CM, Ellebrecht CT, Mukherjee EM, Tang HY, Lin C, Yuan H, Pan M, Langenhan J, Komorowski L, Siegel DL, Payne AS and Stanley JR,
published in
Cell Rep. 2017 Jan 3;18(1):237-247
(PubMed).
These experiments were represented in GPMDB by
64 files, each an individual LC/MS/MS run.
The raw data set was made available through the Massive ProteomeXchange site,
MSV000080368.
It was published as part of the study entitled
"Proteomic Analysis of Pemphigus Autoantibodies Indicates a Larger, More Diverse, and More Dynamic Repertoire than Determined by B Cell Genetics"
by
Chen J, Zheng Q, Hammers CM, Ellebrecht CT, Mukherjee EM, Tang HY, Lin C, Yuan H, Pan M, Langenhan J, Komorowski L, Siegel DL, Payne AS and Stanley JR,
published in
Cell Rep. 2017 Jan 3;18(1):237-247
(PubMed).
These experiments were represented in GPMDB by
64 files, each an individual LC/MS/MS run.
This research was carried out to characterize the antibodies responsible for an autoimmune disease
known as Pemphigus inwhich
antibodies form against the common epidermal protein family the desmogleins.
The experiments involved pull-downs using desmoglein as the bait to obtain samples enriched in anti-desmoglein
antibodies from serum derived from six patients. The results generated small lists of proteins — averaging about 200 per run —
but the results are very complex to interpret, with many immunoglobulin-related sequences with extensive
regions of overlapping tryptic peptides. This data is ideal for anyone interested in developing algorithms
for coping with this type of protein reassembly complexity ("protein inference"). It is also
a good set of data to work through if your main interest is applying proteomics to the immunology of antibody
response.
How Deep is It Really: Mitochondrial Chromosome-Encoded Proteins (2017/1/12)
Last week (see below) it was suggested that cytosolic aminoacyl tRNA synthetases were
a group of proteins that could be used to characterize sampling-related
effects (particularly "under-sampling") in proteomics results sets. This week a group of
proteins that can be used to check the level-of-detection for integral membrane proteins will be
proposed, namely the thirteen protein subunits encoded on the
mitochondrial chromosome.
These thirteen proteins are translated inside of the mitchondrion, using the
mitochrondrial ribosome
(mitoribosome). All of these proteins are inner mitochondrial membrane protein subunits involved in the
electron transport chain
and are required for oxidative phosphorylation. The proteins contain membrane spanning domains and include
some of the most hydrophobic proteins in the human proteome. The members of this group in Homo sapiens are listed
in the Table 1.
These protein subunits are easy to locate in a result list, as they have the only gene names that
begin with "MT-". They have a wide range of observability, ranging from MT-CO2:p
(16,978 ×) to MT-ND4L:p (0 ×). Counting the number of these sequences that are
present in a particular result set obtained from a cell lysate or membrane preparation indicates
of how well an experimental protocol performed for obtaining peptides from integral membrane proteins.
Please note that the observability of
these proteins varies from species to species because of minor changes in the amino acid sequence.
For example, in mice MT-ND4L:p is observable while MT-ND6:p is not.
Interesting data: PXD003818 (2017/1/4)
 The raw data set was made available through the PRIDE ProteomeXchange site,
PXD003818.
It was published as part of the study entitled
"Nuclear Proteomics Uncovers Diurnal Regulatory Landscapes in Mouse Liver"
by
Wang J, Mauvoisin D, Martin E, Atger F, Galindo AN, Dayon L, Sizzano F, Palini A, Kussmann M, Waridel P, Quadroni M, Dulić V, Naef F, and Gachon F,
published in
Cell Metab. 2016 Oct 31. pii: S1550-4131(16)30534-4
(PubMed).
These experiments were represented in GPMDB by
20 result files, each a composite of 12 fractions obtained by off-gel focusing.
The raw data set was made available through the PRIDE ProteomeXchange site,
PXD003818.
It was published as part of the study entitled
"Nuclear Proteomics Uncovers Diurnal Regulatory Landscapes in Mouse Liver"
by
Wang J, Mauvoisin D, Martin E, Atger F, Galindo AN, Dayon L, Sizzano F, Palini A, Kussmann M, Waridel P, Quadroni M, Dulić V, Naef F, and Gachon F,
published in
Cell Metab. 2016 Oct 31. pii: S1550-4131(16)30534-4
(PubMed).
These experiments were represented in GPMDB by
20 result files, each a composite of 12 fractions obtained by off-gel focusing.
These results demonstrate that the experiment succeeded in enriching nuclear proteins from
Mus musculus hepatocytes. They also show that +6 Da lysine SILAC labelling works well
for liver samples from mice fed labelled lysine chow. The mass spectrometry was good quality with good calibration stability over the course
of the multiple fraction measurements. This stability and good parent ion peak shaped
allowed the confident assignment of N and Q deamidations and a significant number of common protein
phosphorylations. The experimental protocol resulted in some urea-generated amine carbamylations (3–4 % of
identifiable peptides) but kept the IAA-generated amine carboxamidomethylations to a minimum (~ 0.2 %).
How Deep is It Really: Cytosolic Aminoacyl tRNA Synthetases (2017/1/2)
For some reason that I have never really understood, journal editors started to allow
the use of the descriptive term "deep" as a qualifier for larger proteomics
result sets. While it has been used frequently in the literature, as far as I know there has
never been any discussion as to how to qualify a data set as being "deep" (or not).
While this term may simply be a reflection of the current trend towards increasingly baroque terms
used to promote a particular group's work, it does suggest an interesting question: How can you
easily state the LOD and the extent of undersampling in a set of results so that it can be readily
explained to biomedical collaborators?
One way to characterize a data set is to compare the proteins observed with a list
of proteins that should be present in a sample. Many groups use this approach, but tend to
be rather coy about the lists of proteins that they use. These lists often are based on the
research interests of the particular group, so they may be difficult to adapt to general
proteomics results. Over the next few weeks, I'll propose a few lists of protein groups
that can be used for specific purposes in proteomics result analysis.
The first of these protein groups is the Cytosolic Aminoacyl tRNA Synthetases. These enzymes are responsible for charging tRNA with
the appropriate amino acid for use in protein synthesis. All of these enzymes must be present for
protein synthesis to occur. Most of these enzymes require only one subunit, with the exception of
Phe-tRNA synthetase which is a heterodimer composed of FASRA:p & FASRB:p. Most of these
enzymes only charge one specific tRNA, with the exceptions EPRS (charges both Glu- and Pro-tRNA)
and SARS (charges both both Ser-tRNA and Sec-rRNA with serine). This enyzme
group is useful for characterizing samples composed mainly of cell contents that were prepared
without affinity purification. The twenty members of this group in Homo sapiens are listed
in the Table 1.
This table shows that the most frequently observed enzyme EPRS:p has been seen a little more than twice as often as the least frequently observed CARS:p
(36,915:16,048), but none of the subunits are inherently difficult to find in MS/MS proteomics data. They
are all mid-sized, soluble cytosolic proteins with many peptides that can be used for identification in either data dependent or
data independent experiments. Simply counting the number of these subunits observed and dividing by 20
gives a very quick estimate of how well an experiment has performed. The higher this value, the less an experiment
has been affected by undersampling.
Tips & Tricks: Trypsin methylation (2016/12/18)
A recent article by M. Schittmayer, et al. — Cleaning out the Litterbox of Proteomic Scientists' Favorite Pet:
Optimized Data Analysis Avoiding Trypsin Artifacts, J Proteome Res. 2016 Apr 1;15(4):1222-9,
PubMed — has stimulated
some interest in testing proteomics data for peptides associated with chemically modified trypsin.
Trypsin can be chemically N-methylated at lysine to reduce the amount of autolytic cleavage, resulting in
enyzmatic activity being sustained over longer periods of time. In practice, the sequencing grade
trypsin offered by ProMega
has become a standard reagent because of this property.
If you want to be sure to catch the modified trypsin peaks in your data using X! Tandem, you should use
the following steps:
P.S. The letter "O" is already used for the rare genomically encoded amino acid pyrolysine (Pyr) and it should never be used in FASTA files to substitute for lysine (K).
Interesting data: PXD002121 (2016/12/13)
 The raw data set was made available through the PRIDE ProteomeXchange site,
PXD002121.
It was published as part of the study entitled
"Sensing Small Changes in Protein Abundance: Stimulation of Caco-2 Cells by Human Whey Proteins"
by
Cundiff JK, McConnell EJ, Lohe KJ, Maria SD, McMahon RJ and Zhang Q,
published in
J Proteome Res. 2016 Jan 4;15(1):125-43
(PubMed).
These experiments were represented in GPMDB by
16 result files.
The raw data set was made available through the PRIDE ProteomeXchange site,
PXD002121.
It was published as part of the study entitled
"Sensing Small Changes in Protein Abundance: Stimulation of Caco-2 Cells by Human Whey Proteins"
by
Cundiff JK, McConnell EJ, Lohe KJ, Maria SD, McMahon RJ and Zhang Q,
published in
J Proteome Res. 2016 Jan 4;15(1):125-43
(PubMed).
These experiments were represented in GPMDB by
16 result files.
While this paper may not be well known, it contains many of the best identifications of human
cellular proteins currently available. The data is composed of 857 RAW files, which have
been organized into 16 multidimensional chromatography runs using 6-plex TMT for relative
quantitation. The experiments were performed using the human male colon adenocarcinoma cell line
CACO-2, which produces significant amounts of L1RE1:p. The only significant experimental artifact was
the commonly found off-target carbamidomethylation of lysine amino groups and peptide N-terminii. It
is an excellent data set to find examplar spectra for peptides derivatized with 6-plex TMT with
HCD fragmentation as well as high accuracy parent & fragment mass determination (Q-Exactive).
Interesting data: PXD003700 (2016/12/8)
 The raw data set was made available through the PRIDE ProteomeXchange site,
PXD003700.
It was published as part of the study entitled
"Proteome-wide analysis of arginine monomethylation reveals widespread occurrence in human cells"
by
Larsen SC, Sylvestersen KB, Mund A, Lyon D, Mullari M, Madsen MV, Daniel JA, Jensen LJ and Nielsen ML,
published in
Sci Signal. 2016 Aug 30;9(443):rs9
(PubMed).
These experiments were represented in GPMDB by
10 result files.
The raw data set was made available through the PRIDE ProteomeXchange site,
PXD003700.
It was published as part of the study entitled
"Proteome-wide analysis of arginine monomethylation reveals widespread occurrence in human cells"
by
Larsen SC, Sylvestersen KB, Mund A, Lyon D, Mullari M, Madsen MV, Daniel JA, Jensen LJ and Nielsen ML,
published in
Sci Signal. 2016 Aug 30;9(443):rs9
(PubMed).
These experiments were represented in GPMDB by
10 result files.
These experiments were undertaken to determine the extent of arginine monomethylation in a normally functioning cellular proteome.
HEK293-T cells were chosen as a stand in for normal cells. The methods used do a good job of enriching for monomethyl-arginine and
the modified residue was easily detectable in the resulting MS/MS data. Dimethyl-arginine was also easily detectable, although in
lesser amounts. The samples were also enriched for the rare PTM hypusine (which only occurs on one residue of EIF5A2:p). The data
makes an excellent case study for testing algorithms attempting to find single amino acid variants (SAVs), as methylation mimics
many common SAVs, which can lead to the over-prediction of SAVs with naïve algorithms.
New Hardware Added to GPMDB (2016/12/7)
GPMDB is the largest source of detailed information about the evidence supporting the observation of proteins, peptides, PTMs and SAVs
using modern tandem mass spectrometry-based proteomics. This means that it has to keep up with the large amount of raw data being made
available through resources like ProteomeXchange, PeptideAtlas, jPOST, Chorus and others. While actually doing the data analysis to convert
the raw data into results can be a bit of a chore, it is something that can be done by simply adding more computers to solve the problems.
Recording that information in the GPMDB datbase is a very linear process that can not be easily parallelized, making this database loading
step a potential bottleneck.
The system GPMDB had been using for the last four years had been optimized repeatedly, but the maximum data recording rate that could be
achieved was about 0.4 million peptides identifications per hour. At this rate, the results generated by analyzing public data were frequently
requiring 24 hour-a-day operation and still there were days when all of the results could not be added: they had to wait for a pause in raw
data availability to complete. This situation has only been getting worse as the size, complexity and tempo of proteomics data set release increases.
To resolve this problem, it was necessary to create a new hardware solution to increase the speed of loading results into GPMDB.
Last week the new hardware was assembled, installed and tested. This new equipment has a proven result loading rate of 5 million
peptide identifications per hour, which gives GPMDB a maximum loading capacity of about 40 billion peptide identifications a year.
This capacity should be sufficient for at least the next three years of efficient operation.
Copyright © 2021, The Global Proteome Machine.
Located at 137 Bannatyne | Privacy Statement
|